
Abstract
Background
In a previous phase 3 trial, treatment with trifluridine–tipiracil (FTD–TPI) prolonged overall survival among patients with metastatic colorectal cancer. Preliminary data from single-group and randomized phase 2 trials suggest that treatment with FTD–TPI in addition to bevacizumab has the potential to extend survival.
Methods
We randomly assigned, in a 1:1 ratio, adult patients who had received no more than two previous chemotherapy regimens for the treatment of advanced colorectal cancer to receive FTD–TPI plus bevacizumab (combination group) or FTD–TPI alone (FTD–TPI group). The primary end point was overall survival. Secondary end points were progression-free survival and safety, including the time to worsening of the Eastern Cooperative Oncology Group (ECOG) performance-status score from 0 or 1 to 2 or more (on a scale from 0 to 5, with higher scores indicating greater disability).
Results
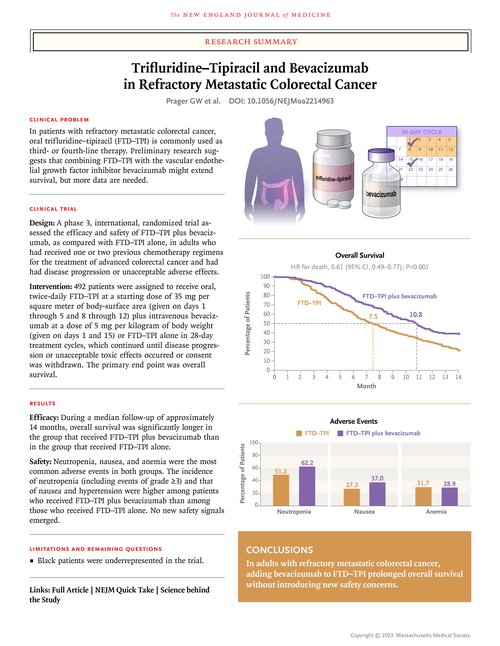
A total of 246 patients were assigned to each group. The median overall survival was 10.8 months in the combination group and 7.5 months in the FTD–TPI group (hazard ratio for death, 0.61; 95% confidence interval [CI], 0.49 to 0.77; P<0.001). The median progression-free survival was 5.6 months in the combination group and 2.4 months in the FTD–TPI group (hazard ratio for disease progression or death, 0.44; 95% CI, 0.36 to 0.54; P<0.001). The most common adverse events in both groups were neutropenia, nausea, and anemia. No treatment-related deaths were reported. The median time to worsening of the ECOG performance-status score from 0 or 1 to 2 or more was 9.3 months in the combination group and 6.3 months in the FTD–TPI group (hazard ratio, 0.54; 95% CI, 0.43 to 0.67).
Conclusions
Among patients with refractory metastatic colorectal cancer, treatment with FTD–TPI plus bevacizumab resulted in longer overall survival than FTD–TPI alone. (Funded by Servier and Taiho Oncology; SUNLIGHT ClinicalTrials.gov number, NCT04737187. opens in new tab; EudraCT number, 2020-001976-14. opens in new tab.)
Patients with metastatic colorectal cancer generally receive first- and second-line treatment with fluorouracil-based chemotherapy (with oxaliplatin and irinotecan), vascular endothelial growth factor (VEGF)–based therapy (mainly bevacizumab), and epidermal growth factor receptor (EGFR)–targeted therapies (the last in patients with RAS wild-type tumors).1,2 Patients who have disease progression after receiving these therapies are considered to have refractory disease; however, many of these patients have a good performance status and may be considered for further therapy.3 Third- and fourth-line treatment options for refractory disease include reintroduction of chemotherapeutic agents such as oxaliplatin,4,5 rechallenge with EGFR therapy in patients with RAS wild-type disease,6-8 and targeted therapies for patients with clinically actionable tumor alterations.9-12 However, most patients receive regorafenib, an oral multikinase inhibitor with antiangiogenic activity, or trifluridine–tipiracil (FTD–TPI).
FTD–TPI is an orally administered combination of trifluridine, a cytotoxic nucleic acid analogue, and tipiracil, a thymidine phosphorylase inhibitor that prevents enzymatic breakdown of trifluridine.13 FTD–TPI was approved as monotherapy for third-line treatment of refractory metastatic colorectal cancer on the basis of the results of the phase 3 RECOURSE trial,13 which showed that overall survival was significantly longer with FTD–TPI therapy than with placebo, irrespective of RAS mutational status,14 and that FTD–TPI had a favorable safety profile. Adverse events were mostly related to myelosuppression.13
Continuous inhibition of angiogenesis beyond progression may be an effective strategy in the management of metastatic colorectal cancer. Maintenance of VEGF inhibition with bevacizumab beyond disease progression has shown clinical activity in patients with metastatic colorectal cancer.15 Furthermore, the results of a randomized, phase 3 trial of regorafenib and a randomized, phase 3 trial of fruquintinib (an inhibitor of VEGF receptors), both of which involved patients with relapsed or refractory disease, showed that overall survival was longer with each of these agents than with best supportive care.16,17 Therefore, combining bevacizumab with FTD–TPI may result in meaningful clinical benefit. Indeed, the combination regimen improved overall survival in several single-group and randomized, phase 2 clinical trials.18-23 The phase 3 SUNLIGHT trial was designed to assess the efficacy and safety of FTD–TPI in combination with bevacizumab as compared with FTD–TPI alone in patients with refractory metastatic colorectal cancer.24
Methods
Patients
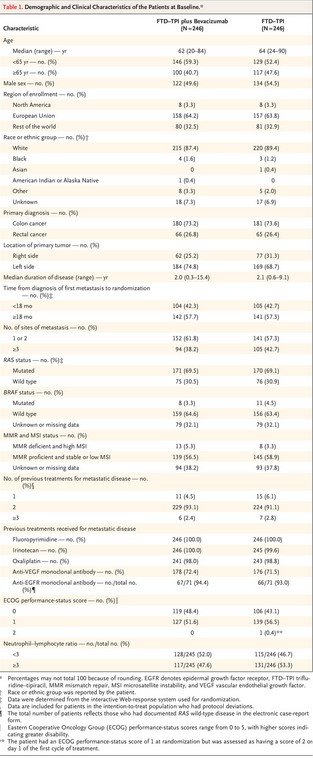
Patients with histologically confirmed, unresectable adenocarcinoma of the colon or rectum were eligible for participation if they had received no more than two previous chemotherapy regimens for the treatment of advanced colorectal cancer and had had progressive disease or if their last regimen had caused unacceptable adverse effects. Previous treatment must have included a fluoropyrimidine, irinotecan, oxaliplatin, an anti-VEGF monoclonal antibody (not necessarily bevacizumab), or an anti-EGFR monoclonal antibody (for patients with RAS wild-type disease), and the treatment could have included neoadjuvant or adjuvant chemotherapy if disease had recurred during treatment or within 6 months after the last administration of neoadjuvant or adjuvant therapy. Eligibility required knowledge of RAS status. Patients had to be 18 years of age or older and have adequate organ function and an Eastern Cooperative Oncology Group (ECOG) performance-status score of 0 or 1 (with scores ranging from 0 to 5 and higher scores indicating greater disability).
Trial Design and Treatments
Patients were randomly assigned in a 1:1 ratio to receive FTD–TPI (Lonsurf, Servier and Taiho Oncology) plus bevacizumab (Avastin, Genentech and Roche) (combination group) or FTD–TPI alone (FTD–TPI group). Randomization was stratified according to geographic region (North America, European Union, or rest of the world), time since diagnosis of first metastasis (<18 months or ≥18 months), and RAS status (wild type or mutated).
FTD–TPI was administered orally, twice daily, at a starting dose of 35 mg per square meter of body-surface area, on days 1 through 5 and on days 8 through 12 every 28 days. Bevacizumab, at a dose of 5 mg per kilogram of body weight, was administered intravenously on days 1 and 15. The 28-day treatment cycle continued until disease progression or unacceptable toxic effects occurred or consent was withdrawn. Patients were considered to be receiving treatment for as long as they continued to receive FTD–TPI; bevacizumab monotherapy was not allowed.
End Points and Assessments
The primary end point was overall survival, defined as the time from randomization to death from any cause. Secondary end points included investigator-assessed progression-free survival; objective response and disease control according to Response Evaluation Criteria in Solid Tumors, version 1.125; quality of life, assessed with the use of the patient-completed European Organization for Research and Treatment of Cancer Quality-of-Life Questionnaire–Core 30, version 3.0, and EuroQol 5-Dimension 5-Level questionnaire (quality-of-life data are not reported here); and safety, which included the assessment of adverse events, laboratory tests, physical examinations, vital signs, and the time from randomization to worsening of the ECOG performance-status score from 0 or 1 to 2 or more.
Efficacy was assessed in all the patients who had undergone randomization, in accordance with the intention-to-treat principle. Safety was assessed in all the patients who received one or more doses of a trial agent.
Tumor imaging was performed at baseline and every two cycles until progression was observed on imaging. Patients were contacted to assess vital status every 8 weeks during follow-up until death occurred or until the end of the trial. Data on adverse events and laboratory abnormalities were collected regularly throughout the treatment period and for 30 days thereafter. Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0.
Trial Oversight and Conduct
The trial was sponsored by Servier and Taiho Oncology and designed by the last author and representatives of Servier. The trial protocol (available with the full text of this article at NEJM.org) was approved by the institutional review board or ethics committee at each trial site before the start of the trial, in accordance with local regulations. All the patients provided written informed consent before enrollment. The trial was performed in accordance with the principles of the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Council for Harmonisation. An independent data and safety monitoring board regularly reviewed and evaluated the conduct and safety of the trial. All the authors participated in the collection, analysis, and interpretation of the data. The sponsors were involved in the design and conduct of the trial, the collection and analysis of the data, the writing of the manuscript, and the decision to submit the manuscript for publication. All the authors vouch for the accuracy and completeness of the data and for the fidelity of the trial to the protocol, attest that they had access to the data and participated in reviewing and editing earlier drafts of the manuscript, and agreed to submit the manuscript for publication. The first draft of the manuscript was written by a medical writer, funded by the sponsors. Data confidentiality agreements were in place between the authors and the sponsors.
Statistical Analysis
The primary objective was to show the superiority of FTD–TPI plus bevacizumab over FTD–TPI alone with respect to overall survival. The trial was designed to have 90% power to detect a hazard ratio of 0.70 (a 30% lower risk of death during the observation period with FTD–TPI plus bevacizumab than with FTD–TPI alone), with the use of a log-rank test and a one-sided type I error rate of 0.025. A total of 490 patients (245 in each group) and at least 331 events (death from any cause) were required for the primary analysis. A hierarchical testing strategy was used to control the overall type I error rate; progression-free survival would be evaluated only if the primary analysis showed that overall survival differed significantly between the two trial groups. Overall survival and progression-free survival reflected the duration of survival in all patients, regardless of whether an intercurrent event (defined as the administration of additional anticancer therapy, treatment discontinuation, or a switch between trial groups) occurred. A stratified log-rank test at a two-sided 5% significance level was used to compare the distributions of overall survival and progression-free survival between the two trial groups, and a stratified Cox proportional-hazards model was used to assess the magnitude of the treatment difference.
Subgroup analyses of overall survival and progression-free survival were prespecified to assess the homogeneity of the treatment effect across subgroups of patients. An unstratified Cox-regression model with trial group as a predictor variable was fitted separately for each subgroup category, and the hazard ratio for the assigned treatment, along with the associated 95% confidence interval, was determined. A prespecified multivariate analysis of overall survival was also performed with the use of a Cox proportional-hazards model; variables were identified for inclusion in a multivariable model by means of stepwise selection on the basis of P values. Because choosing variables in this fashion can result in omission of important confounders and underestimation of the widths of confidence intervals, an additional multivariable-adjusted analysis of overall survival, including all proposed potential confounders without stepwise variable selection, was performed as an ad hoc analysis. Two-sided 95% Clopper–Pearson confidence intervals were used to describe objective response and disease control in each trial group. A two-sided 95% confidence interval for the between-group difference in these outcomes was provided on the basis of normal approximation.
Safety data were summarized with the use of descriptive statistics. The time from randomization to worsening of the ECOG performance-status score from 0 or 1 to 2 or more or death was analyzed with the use of the Kaplan–Meier method, and a stratified Cox proportional-hazards model was used to assess the magnitude of treatment difference.
The stratification factors used at randomization were applied to all stratified analyses. For all analyses, the widths of the confidence intervals were not adjusted for multiplicity and may not be used in place of hypothesis testing. Additional methods are described in the Supplementary Appendix, available at NEJM.org.
Results
Patients
Figure 1. Randomization and Treatment.Table 1. Demographic and Clinical Characteristics of the Patients at Baseline.
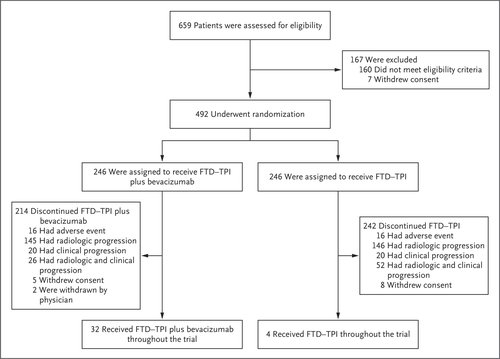
Overall, 659 patients at 87 sites in 13 countries were screened for eligibility; of these, 492 patients underwent randomization from November 25, 2020, to February 18, 2022. A total of 246 patients were assigned to each group (Figure 1). The demographic and clinical characteristics of the patients at baseline were balanced between the two groups (Table 1). Most patients (64.0%) were from the European Union. The time from the diagnosis of the first metastasis until randomization was 18 months or longer in 57.5% of the patients, and 30.7% had RAS wild-type disease.
Most patients (92.1%) had received two previous treatment regimens for metastatic disease; however, 4.5% of the patients in the combination group and 6.1% in the FTD–TPI group had received only one first-line triplet regimen, and 2.6% of the patients in the trial had received three or more previous drug regimens for metastatic disease. All the patients had received previous fluoropyrimidine-based therapy, 72.0% had received previous anti-VEGF therapy (47.8% had received bevacizumab as part of their first regimen, 43.9% as part of their second regimen, and 20.3% as part of both their first and second regimens), and 93.7% of the patients with RAS wild-type disease had received previous anti-EGFR therapy (Table 1).
The trial population was largely representative of the expected patient population (Table S1 in the Supplementary Appendix), except that the percentage of Black patients was relatively low, owing to recruitment of patients from predominantly White countries. The percentage of patients with RAS mutations (69.3%) was higher than that in the general population of patients with metastatic colorectal cancer, potentially reflecting preferential referral of patients with RAS wild-type tumors to clinical trials of anti-EGFR therapy.
The median duration of treatment was 5.0 months (range, 0.1 to 18.5) in the combination group and 2.1 months (range, 0.6 to 14.3) in the FTD–TPI group. In the combination group, the median relative dose intensities of FTD–TPI and bevacizumab were 88.3% and 87.6%, respectively. The median relative dose intensity of FTD–TPI in the FTD–TPI group was 90.4%.
At the time of the analysis, 13.0% of the patients in the combination group and 1.6% of the patients in the FTD–TPI group were still receiving treatment. The most common reason for treatment discontinuation was disease progression (Figure 1). Overall, treatment was discontinued in six patients owing to the coronavirus disease 2019 pandemic.
Efficacy
Figure 2. Overall Survival and Progression-free Survival (Intention-to-Treat Population).
The median follow-up was 14.2 months (interquartile range, 12.6 to 16.4) in the combination group and 13.6 months (interquartile range, 12.7 to 15.9) in the FTD–TPI group. The median overall survival was 10.8 months (95% confidence interval [CI], 9.4 to 11.8) in the combination group and 7.5 months (95% CI, 6.3 to 8.6) in the FTD–TPI group (hazard ratio for death, 0.61; 95% CI, 0.49 to 0.77; P<0.001) (Figure 2A). A sensitivity analysis that excluded patients who did not meet relevant prespecified medical and therapeutic criteria (14 patients in the combination group and 16 patients in the FTD–TPI group) showed a median overall survival of 10.8 months (95% CI, 9.6 to 12.1) in the combination group and 7.2 months (95% CI, 6.3 to 8.5) in the FTD–TPI group (hazard ratio for death, 0.59; 95% CI, 0.47 to 0.74). The multivariate model estimate of treatment effect (FTD–TPI plus bevacizumab relative to FTD–TPI) was consistent with that of the primary analysis (hazard ratio for death, 0.63; 95% CI, 0.50 to 0.78).
The 6-month overall survival was 77% in the combination group and 61% in the FTD–TPI group; the 12-month overall survival was 43% and 30%, respectively. The median progression-free survival was 5.6 months (95% CI, 4.5 to 5.9) in the combination group and 2.4 months (95% CI, 2.1 to 3.2) in the FTD–TPI group (hazard ratio for disease progression or death, 0.44; 95% CI, 0.36 to 0.54; P<0.001). The 6-month progression-free survival was 43% in the combination group and 16% in the FTD–TPI group; the 12-month progression-free survival was 16% and 1%, respectively (Figure 2B). The benefits of FTD–TPI plus bevacizumab with respect to overall survival and progression-free survival were observed in all subgroups examined (Figs. S1 and S2). An objective response was observed in 6.1% of the patients in the combination group (95% CI, 3.5 to 9.9), with 15 patients having had a partial response, and in 1.2% of the patients in the FTD–TPI group (95% CI, 0.3 to 3.5), with 1 patient having had a complete response and 2 patients having had a partial response (between-group difference, 4.9 percentage points; 95% CI, 1.6 to 8.2).
Safety
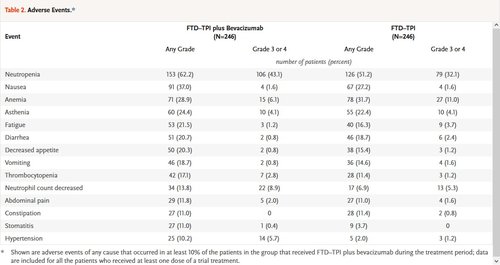
Table 2. Adverse Events.
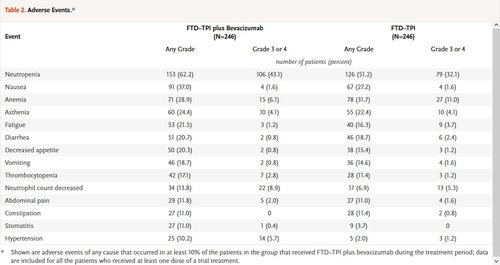
Adverse events of any cause occurred in 98.0% of the patients in each group. Severe adverse events (grade ≥3) were reported in 72.4% of the patients in the combination group and in 69.5% of the patients in the FTD–TPI group. Serious adverse events were reported in 24.8% and 31.3% in the two groups, respectively. The most common adverse events that occurred during the treatment period in both groups were neutropenia, nausea, and anemia (Table 2). The events that were more frequent in the combination group than in the FTD–TPI group were hypertension (in 10.2% of the patients in the combination group and 2.0% in the FTD–TPI group), nausea (in 37.0% and 27.2%, respectively), and neutropenia (in 62.2% and 51.2%, respectively), including severe (grade ≥3) neutropenia (in 43.1% and 32.1%, respectively). Concomitant granulocyte colony-stimulating factors were administered in 29.3% of the patients in the combination group and in 19.5% in the FTD–TPI group during the treatment period. Overall, 89.8% of the patients in the combination group and 81.3% in the FTD–TPI group had adverse events that were attributed by the investigator to FTD–TPI, and 48.4% of the patients in the combination group had bevacizumab-related events. No treatment-related deaths were reported.
Adverse events of any cause that led to discontinuation of the trial regimen were reported in 12.6% of the patients in both the combination group and the FTD–TPI group. Of these events, the investigators determined that the events were related to treatment in 2.4% and 2.0%, respectively. Dose reductions occurred in 16.3% of the patients in the combination group and in 12.2% in the FTD–TPI group; dose delays occurred in 69.5% and 53.3%, respectively. The median time to worsening of the ECOG performance-status score from 0 or 1 to 2 or more was 9.3 months (95% CI, 8.3 to 10.6) in the combination group and 6.3 months (95% CI, 5.6 to 7.2) in the FTD–TPI group (hazard ratio, 0.54; 95% CI, 0.43 to 0.67).
Discussion
This phase 3, international, prospective, randomized, active-controlled, trial involving patients with refractory metastatic colorectal cancer showed that treatment with FTD–TPI plus bevacizumab resulted in significantly longer overall survival and progression-free survival and better disease control than treatment with FTD–TPI alone. Although a minority of patients did not receive the trial treatment as third-line therapy, this trial was predominantly a third-line trial (>90% of patients had received two previous lines of therapy). The duration of overall survival in the FTD–TPI group was consistent with previous observations,13,26 a finding that suggests the benefits observed with FTD–TPI plus bevacizumab will be applicable to all suitable patients with refractory disease.
The survival benefits of FTD–TPI plus bevacizumab were observed in all subgroups, including the subgroup of patients with factors indicative of a poor prognosis.14 Survival benefits of the combination were observed irrespective of age, sex, location of primary disease, number of metastatic sites, or RAS mutation status, a finding that indicates FTD–TPI plus bevacizumab is an option for all clinically relevant subgroups. Clinical benefit was also observed regardless of whether patients had received previous treatment with bevacizumab, a finding that adds to the body of evidence supporting a role for continued inhibition of angiogenesis beyond progression15 and suggesting that the addition of bevacizumab to third-line or later-line therapy may prolong survival in heavily pretreated patients.
The results of this trial are noteworthy for several reasons. First, the trial showed superior efficacy of a regimen to an existing proven therapy. Second, bevacizumab has not previously shown substantial efficacy beyond second-line treatment in trials of metastatic colorectal cancer.27 Third, 69% of the patients in this trial had tumors with a RAS mutation, a percentage that is higher than that generally reported in the population of patients with metastatic colorectal cancer.28 This is notable because in randomized trials, bevacizumab plus chemotherapy has been reported to provide only marginal overall survival benefits in patients with RAS-mutated colorectal cancer.29 Fourth, the overall survival benefit observed in this trial (hazard ratio for death, 0.61) is larger than the magnitude of benefit observed in other bevacizumab-based combination trials with active controls, even as first-line therapy.30 Therefore, these results in patients with refractory disease are encouraging.
The addition of bevacizumab to FTD–TPI did not increase the risk of serious adverse events or adverse events leading to treatment discontinuation. As previously observed,31-33 FTD–TPI plus bevacizumab was associated with a higher incidence of severe neutropenia than FTD–TPI alone, possibly related to an increased accumulation of phosphorylated trifluridine, facilitated by bevacizumab.34 However, no increase in the incidence of febrile neutropenia was seen with the combination regimen (febrile neutropenia occurred in one patient in the combination group and in six patients in the FTD–TPI group). Dose modifications were also more common in the combination group than in the FTD–TPI group; however, this was anticipated, since patients in the former group were assessed more frequently owing to the treatment schedule. The time to worsening of the ECOG performance-status score from 0 or 1 to 2 or more was longer among patients in the combination group than among patients in the FTD–TPI group; this finding is important because prolonging physical performance and controlling symptoms may allow patients to maintain their physical function and further benefit from subsequent therapy.
FTD–TPI plus bevacizumab showed better survival outcomes than FTD–TPI alone in most prespecified subgroups of patients. The safety profile of FTD–TPI plus bevacizumab was as expected, and patients who received the combination had longer preservation of performance status than patients who received FTD–TPI alone. The data from this trial confirm that FTD–TPI plus bevacizumab is an effective treatment option for patients with refractory metastatic colorectal cancer, irrespective of mutational status, which side the tumor is on, and whether patients have previously been treated with bevacizumab.
Author Affiliations
From the Department of Medicine I, Comprehensive Cancer Center, Medical University of Vienna, Vienna (G.W.P.); the Department of Gastroenterology and Digestive Oncology, Georges Pompidou European Hospital, SIRIC Cancer Research for Personalized Medicine, Université Paris Cité, Paris (J. Taieb), the Department of Oncology, University Hospital, Brest (P.-G.P.), and Servier, Suresnes (N.A., C.L., L.V.) — all in France; City of Hope Comprehensive Cancer Center, Duarte, CA (M.F.); Dipartimento di Medicina di Precisione, Università degli Studi della Campania Luigi Vanvitelli, Naples (F.C.), and the Unit of Medical Oncology, Department of Translational Research on New Technologies in Medicine and Surgery, University of Pisa, Pisa (C.C.) — both in Italy; the Department of Digestive Oncology, University Hospitals Gasthuisberg and KU Leuven, Leuven, Belgium (E.V.C.); the Department of Medical Oncology, Vall d’Hebron Hospital Campus and Institute of Oncology, International Oncology Bureau–Quiron, Barcelona (E.E., J. Tabernero); Núcleo de Pesquisa e Ensino da Rede São Camilo, São Paulo (F.M.C.); the Department of Oncology and Radiotherapy, Maria Sklodowska-Curie National Research Institute of Oncology, Warsaw, Poland (L.W.); Moscow City Oncology Hospital, Moscow Healthcare Department, Moscow (D.S.); the Department of Oncology, Hungarian Defense Forces Medical Center, Budapest, Hungary (Z.P.); the Department of Oncology, Regional Hospital West Jutland, Herning, Denmark (G.L.); Dnipro State Medical University, Dnipro, Ukraine (I.B.); the Medical Department of Hematology, Oncology, and Cancer Immunology, Charité–Universitätsmedizin Berlin, Berlin (D.P.M.); and Taiho Oncology, Princeton, NJ (K.A.B.).
Dr. Tabernero can be contacted at jtabernero@vhio.net or at the Vall d’Hebron Barcelona Hospital Campus, Passeig de la Vall d’Hebron 119-129, 08035 Barcelona, Spain.
A list of the SUNLIGHT Investigators is provided in the Supplementary Appendix, available at NEJM.org.

